

 Terzo appuntamento con le malattie di cui si parlerà nel convegno Bio.Med.Day: la scienza medica e le sue prospettive future, organizzato dall’Unione degli Universitari – Sinistra Universitaria (Udu) e Udu Medicina, anche in collaborazione con TESSERE, in programma per martedì 6 marzo dalle ore 9 alle 19 al Plesso didattico Morgagni di Firenze. Dopo gli articoli Alla scoperta dei misteri racchiusi nel cuore, e Leucemia: la speranza è nella terapia genica, oggi si affronta una grave patologia neurodegenerativa, il Morbo di Huntington. Al convegno interverranno: Elena Cattaneo, direttrice del laboratorio di Biologia delle cellule staminali e Farmacologia delle malattie neurodegenerative dell’Università Statale di Milano; Michele De Luca, direttore del Centro di Medicina rigenerativa dell’Università di Modena e Reggio Emilia e co-presidente dell’Associazione Luca Coscioni; Silvia G. Priori, professore associato presso l’Università degli Studi di Pavia, primario dell’Unità operativa di Cardiologia molecolare presso la Fondazione Maugeri di Pavia e direttrice del Dipartimento di Genetica cardiovascolare alla University School of Medicine di New York; Giuseppe Remuzzi, direttore dell’Unità operativa di Nefrologia e dialisi e del Dipartimento di Medicina dell’Azienda ospedaliera Papa Giovanni XXIII di Bergamo. Gli articoli, scritti da studenti universitari delle facoltà medico-scientifiche dell’Università di Firenze, verranno pubblicati anche nel giornale realizzato dagli studenti di Udu Medicina.
Terzo appuntamento con le malattie di cui si parlerà nel convegno Bio.Med.Day: la scienza medica e le sue prospettive future, organizzato dall’Unione degli Universitari – Sinistra Universitaria (Udu) e Udu Medicina, anche in collaborazione con TESSERE, in programma per martedì 6 marzo dalle ore 9 alle 19 al Plesso didattico Morgagni di Firenze. Dopo gli articoli Alla scoperta dei misteri racchiusi nel cuore, e Leucemia: la speranza è nella terapia genica, oggi si affronta una grave patologia neurodegenerativa, il Morbo di Huntington. Al convegno interverranno: Elena Cattaneo, direttrice del laboratorio di Biologia delle cellule staminali e Farmacologia delle malattie neurodegenerative dell’Università Statale di Milano; Michele De Luca, direttore del Centro di Medicina rigenerativa dell’Università di Modena e Reggio Emilia e co-presidente dell’Associazione Luca Coscioni; Silvia G. Priori, professore associato presso l’Università degli Studi di Pavia, primario dell’Unità operativa di Cardiologia molecolare presso la Fondazione Maugeri di Pavia e direttrice del Dipartimento di Genetica cardiovascolare alla University School of Medicine di New York; Giuseppe Remuzzi, direttore dell’Unità operativa di Nefrologia e dialisi e del Dipartimento di Medicina dell’Azienda ospedaliera Papa Giovanni XXIII di Bergamo. Gli articoli, scritti da studenti universitari delle facoltà medico-scientifiche dell’Università di Firenze, verranno pubblicati anche nel giornale realizzato dagli studenti di Udu Medicina.
La malattia, o Morbo di Huntington è una rara patologia neurodegenerativa classificata sotto la categoria delle Còree. Queste sono patologie che, col passare del tempo e dunque la loro progressione, portano nel malato disturbi cognitivi, della memoria e come manifestazione tipica, a compiere movimenti scoordinati, privi di senso che possono anche sembrare un ballo, una sorta di danza logorante che rende impossibili le attività quotidiane; anche un semplice gesto come un saluto, una carezza, avvicinare il cibo alla bocca può trasformarsi in una ripetizione insensata e spasmodica di movimenti.
Sebbene la malattia venga spesso confusa e mitizzata come “Ballo di San Vito”, questo nome è appropriato per una Còrea diversa, conosciuta (come anche la corea di Huntington) sin dal medioevo, quella di Sydenham. Entrambe le malattie sono state poco studiate nella storia fino agli ultimi decenni del XX secolo, complice di questo l’eziologia strettamente legata alla Genetica, che solo in tempi recenti ha consentito agli addetti ai lavori di comprendere qualcosa in più sulla sua genesi e trasmissione.
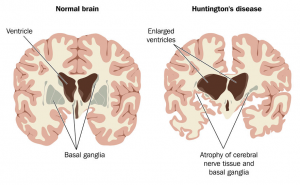 L’Huntington infatti è uno di quei “disordini in triplette”, malattie legate a proteine che vengono codificate nel genoma umano attraverso dei codoni ripetuti nel DNA. Nella maggior parte delle persone, all’interno del cromosoma 4, sono contenute dalle 10 alle 35 ripetizioni della tripletta “CAG” che formano il gene IT15, codificante per l’Huntingtina, proteina formata da tante glutammine quante sono queste triplette.
L’Huntington infatti è uno di quei “disordini in triplette”, malattie legate a proteine che vengono codificate nel genoma umano attraverso dei codoni ripetuti nel DNA. Nella maggior parte delle persone, all’interno del cromosoma 4, sono contenute dalle 10 alle 35 ripetizioni della tripletta “CAG” che formano il gene IT15, codificante per l’Huntingtina, proteina formata da tante glutammine quante sono queste triplette.
Nelle persone affette da Còrea il numero di ripetizioni è maggiore della norma, dalle 36 triplette in poi si evidenziano differenze nel funzionamento di alcune popolazioni di neuroni che si trovano in agglomerati di materia grigia detti “nuclei della base”.
Il meccanismo non è ancora chiaro, si sa però che questi nuclei sono coinvolti nella produzione dei movimenti volontari, sono come dei circuiti, definiti dagli anatomisti vie extrapiramidali, che appaiono attivi negli elettroencefalogrammi quando un soggetto sta per effettuare un movimento volontario. Il collegamento è diretto, tuttavia è difficile comprendere appieno il meccanismo molecolare per correggere i sintomi della malattia.
Le terapie applicate in questo momento sono per lo più concentrate sull’alleviare i sintomi, spesso con piccoli risultati che si manifestano più come una sedazione del paziente; per dare un esempio conosciuto a molti, i pazienti vengono trattati con massicce dosi di benzodiazepine, le stesse molecole contenute nei farmaci ansiolitici più comuni (es. Alprazolam, meglio conosciuto come Xanax), oppure antidepressivi e ancora modulatori della serotonina che non solo non hanno nessun effetto sulla causa della malattia o sul rallentare il suo progredire ma anzi, causano assuefazione e talvolta possono peggiorare lo stato psicologico del malato.
 Le strategie del domani mirano ad una terapia a livello genico o attraverso l’uso di cellule staminali, ed estremamente importante in questo senso è la ricerca della professoressa Elena Cattaneo, docente di farmacologia nel dipartimento di bioscienze dell’università di Milano e senatrice a vita. Da più di vent’anni si occupa di Huntington non solo dal punto di vista scientifico ma anche dal punto di vista umano. Nei suoi laboratori si tenta di trovare una cura per la malattia che sembra convergere verso l’uso delle staminali.
Le strategie del domani mirano ad una terapia a livello genico o attraverso l’uso di cellule staminali, ed estremamente importante in questo senso è la ricerca della professoressa Elena Cattaneo, docente di farmacologia nel dipartimento di bioscienze dell’università di Milano e senatrice a vita. Da più di vent’anni si occupa di Huntington non solo dal punto di vista scientifico ma anche dal punto di vista umano. Nei suoi laboratori si tenta di trovare una cura per la malattia che sembra convergere verso l’uso delle staminali.
Queste cellule, nel momento in cui possiedono maggiori proprietà di pluripotenza, la capacità di decidere di essere cellule della pelle o del cervello, si trovano soltanto in un piccolissimo intervallo temporale dello sviluppo di un individuo umano; questo è il periodo in cui l’embrione è allo stadio di blastocisti, un periodo sul quale è difficile lavorare anche per le sue implicazioni etiche.
Nell’andare avanti le cellule sempre di più si specializzano e sono legate ad un destino funzionale specifico. Ogni tessuto mantiene però un certo numero di staminali, non duttili come quelle embrionali ma sulle quali è possibile lavorare. Sono state dunque identificate delle catene di cellule capaci di generare nuovi neuroni nell’arco di 4 settimane anche all’interno del cervello adulto, nella zona dell’ippocampo e sottoventricolare, attraverso le quali si studia la cura per le malattie neurodegenerative come l’Huntington.
Questa materia richiede tuttavia un approccio integrato non solo nel campo delle biotecnologie e della medicina ma anche nel campo della filosofia della scienza, della bioetica.
 Dalle ricerche della professoressa Cattaneo sul processo di formazione delle cellule nel sistema nervoso in età embrionale, sono emersi dati che collegano le stesse proteine coinvolte nel morbo di Huntington alla formazione di complessi a forma di “fiore” chiamate rosette neurali strettamente legati all’evoluzione del sistema nervoso nella specie umana.
Dalle ricerche della professoressa Cattaneo sul processo di formazione delle cellule nel sistema nervoso in età embrionale, sono emersi dati che collegano le stesse proteine coinvolte nel morbo di Huntington alla formazione di complessi a forma di “fiore” chiamate rosette neurali strettamente legati all’evoluzione del sistema nervoso nella specie umana.
Il numero delle stesse triplette patogenetiche ha una tendenza ad essere maggiore nelle specie più in alto nella scala evolutiva, configurando così un complesso paradosso biologico, malattia-evoluzione, da studiare ancora a fondo.
Anche se i risultati sono ancora troppo eterogenei per descrivere linee guida precise nel trattamento di certe malattie con queste nuove tecniche, si parla di una medicina di tipo Rigenerativo che fa sperare in approcci di cura risolutivi per malattie come l’Huntington o il Parkinson e, sebbene bisogni frenare le aspettative verso questo tipo di trattamenti, la direzione in cui si muove la ricerca sembra ad ogni scoperta più chiara.


